流行病学
流行病学
流行病学:血栓性血小板减少性紫癜(Thrombotic Thrombocytic Purpura,TTP)是一种突然发作的威胁生命的疾病,该病临床具有典型的五联征:发热、血小板减少、溶血、肾功能障碍、神经系统症状。最先由Moschcowitz于1924年报道,其后这类疾病的报道逐渐增多。1947年Singer总结了以前的病例,提出上述五联征作为该病的特征。由于广泛的微血管病变,该病的临床表现复杂(表1)。本病主要分为两型:急性获得型和相对罕见的遗传性慢性反复发作型。急性型又可以进一步分为特发性和继发性。药物、感染以及其他一些因素与继发性TTP有关。国外文献报道,年发病率约为3/100万。近年由于对该病认识的加深,继发于其他疾病和药物的患者增多,估计实际发病率可能更高。发病情况通常与种族差异无关,女性多于男性,二者比为3∶2。虽然婴儿和90岁以上老年人均可发病,但是发病的高峰年龄是20~60岁,中位年龄35岁。总体看来,本病在儿童还是相对少见。
病因
病因:本病征病因未明,主要是由于小血管病变,可有毒素、药物过敏、细菌或病毒感染、自身免疫、胶原性疾病等学说,但均未能证实。常见于伴有小血管病变的疾病,如红斑性狼疮、多发性动脉炎、舍格伦综合征和类风湿性关节炎等。由于小血管病变,微血管的弥漫性病变,使血管内溶血促进
血栓形成。
发病机制
发病机制:
1.病理生理 vWF是正常止血过程中必需的成分,在高剪切力血流状态时内皮细胞表面、血小板表面受体和vWF多聚体三者之间就会相互作用,导致血小板与内皮细胞黏附。vWF缺乏或分子结构异常会导致一种较常见的遗传性出血性疾病――血管性血友病(vWD)。另一方面,vWF水平过高会造成慢性内皮细胞损伤,可导致血栓性疾病。vWF被分泌到血浆后要经历酶解过程,酶切位点是其A2结构域的Tyr842-Met843,执行这一酶切作用的是vWF切割蛋白(Vwf cleavage protein,vWFCP),亦即凝血酶敏感蛋白1基序的裂解素和金属蛋白酶家族成员13(adisintegrin and metalloproteinase with thrombospodin type 1 motif 13,ADAMTS13)。vWF多聚体越大,止血活性越强。在复发性TTP患者易出现超大分子量的vWF多聚体,在高剪切力情况下与血小板结合能力要比平时强的多,从而形成广泛的微血栓。Moake等认为超大vWF多聚体的出现可能是由于内皮细胞过度分泌vWF或降解vWF的酶系统缺乏所致,这一推测为后来的研究所证实。1996年,Furlan和Tsai分别证明在二价金属离子条件下一种金属蛋白酶切割vWF,其缺乏可导致超大vWF多聚体形成,这种金属蛋白酶即vWFCP。以后的研究进一步证实vWFCP结构缺陷与家族性TTP密切相关,而后天获得性vWFCP自身抗体则会造成非家族性TTP。
内皮细胞损伤被认为是TTP发病机制中关键的启动因素,可导致血小板聚集并在脑、肾和其他器官微血管内形成血小板血栓。一般来说,正常状态下微血管内皮是抗凝的。但是一旦被破坏或激活,内皮细胞将丧失抗凝活性而变成促凝物。Hamilton等发现在暴露于补体复合物C5b-9几分钟内人脐静脉内皮细胞可释放内皮细胞微粒(Endothial micropaiticles,EMPs),在TNFa存在24~48h内EMPs可表达组织因子、结构抗原CD31和CD51,并且体现了促凝活性。Jimenez等发现TTP患者血浆可诱导脑和肾的EMPs水平增高5倍,促凝活性增高2~3倍,并且使细胞间黏附分子(ICAM-1)表达增高3倍,VCAM-1增高13倍,而对照组血浆和ITP患者的血浆则无此作用,表明TTP发生可能主要与内皮细胞激活有关。临床发现在TTP急性期EMP水平显著增高,恢复期时EMP水平与对照组相当。因此他们推论从EMP水平及激活标记物表达来看,TTP患者血浆可激活并诱导微血管内皮细胞损伤。最近他们以TTP患者的血浆诱导微血管内皮细胞发现释放出来的EMP高表达激活相关标志CD62E和CD54,进一步支持内皮细胞激活在TTP发病学的作用。
促凝物EMP的释放可能在TTP中有主要作用。EMP可能是一种有前景的判断TTP甚至其他血栓性疾病激活和内皮损伤的标志物。EMP可能触发或扩大血小板微血栓的形成。对EMP的深入研究有助于深化TTP或血栓性微血管病的认识。
2.病理特点 TTP属于微血管病,全身微动脉和毛细血管均可受累。过度增生的内皮细胞和血小板性微血栓堵塞了血管,这种不完全的堵塞造成红细胞在通过微血管时损伤形成碎片细胞(裂细胞)。光镜下微血栓呈颗粒状,PAS和GIEMSA染色阳性,免疫荧光和电镜研究均证明这种血栓主要成分是纤维素和血小板,偶尔也能发现补体和免疫球蛋白。由于内皮细胞过度增生,导致有些血栓出现在内皮下。虽然不像通常的炎症病理那样有炎细胞浸润,但是受累血管的内皮细胞还是有形态学和功能改变。电镜下这些细胞肿胀,胞质内像微管样的纤维素成分很多。在血小板聚集区域的内皮细胞包含有大量的胞质成分,内质网表面粗糙,线粒体增多,高尔基区变大,还有大量的溶酶体。
血管损伤部位极为广泛,没有特异性,但是以肾脏、脑、脾、胰腺、心脏和肾上腺等部位最为常见,尤其是来源于心、肾、脑的内皮细胞更容易被体内的TTP血浆损伤。皮肤、牙龈和骨髓活检可以发现30%~50%的患者有损伤证据。最常见的出血部位是受损血管附近,这些地方表现为紫癜,是皮肤活检的理想部位。虽然胰腺和肾脏出血和栓塞最常见,但是最广泛的出血经常发生在脑部,从而导致更严重的结果。
临床表现
临床表现:多数TTP患者既往身体健康(特发性TTP),大约15%左右的患者可以继发于某些疾病,这类患者称之为继发性TTP,这些常见的继发原因有细菌或病毒感染、怀孕、胶原血管疾病或者胰腺炎等。某些药物也可以诱发TTP-HUS,例如氯吡格雷、奎宁、丝裂霉素C、环孢霉素以及FK506(免疫抑制药)等。这些药物导致TTP的发病机制尚不明确,不过已经发现在某些服用过氯吡格雷相关的TTP患者可以检测到ADAMTS13的抑制性抗体。
多数情况下,TTP起病比较突然,最常见的首发症状是出血和神经系统异常表现。神经症状迅速进展可以出现
头痛、困倦、木僵、
昏迷、脑神经
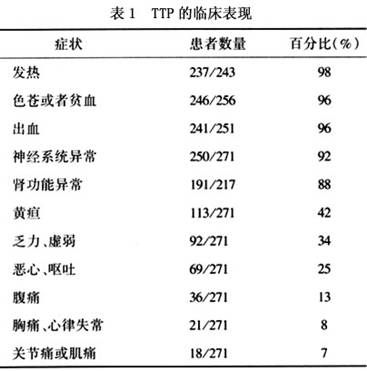
麻痹和癫痫发作等。发热在本病开始时并不一定出现,随着病情的进展可以逐渐出现(表1)。
1.发热 常在38~40℃,90%以上患者有发热,发热原因可由于病变侵犯下丘脑、组织坏死、溶血物质释放、抗原抗体反应以及并发感染。
2.出血 主要是血小板减少,紫癜及视网膜出血最常见,其次是胃肠道及泌尿道等。
3.
溶血性贫血 由于毛细血管内纤维蛋白沉着,使管腔狭窄,压挤红细胞使之破碎,因溶血可出现不同程度的
黄疸。
4.神经系统受累的症状 脑血管被侵害,出现多种多样、时轻时重、暂时性或永久性的神经症状。
5.肾脏病变 可见肾小管透明栓塞,甚至肾皮质坏死而引起
急性肾功能衰竭。
并发症
实验室检查
实验室检查:
1.贫血 常较严重,为正色素性贫血,网织红细胞及
有核红细胞明显增高,红细胞形态异常和破碎,骨髓红系统增生活跃,
红细胞寿命缩短为3~6天,血清胆红素增高呈间接反应。
2.白细胞增高 部分病例呈类白血病反应。
3.血小板 血小板减少,骨髓巨核细胞正常。
其他辅助检查
其他辅助检查:血管脆性试验常为阳性,应做各种影像学检查,如胸片、胃肠钡剂造影、B超、脑CT及脑电图等检查。
诊断
诊断:根据临床溶血、贫血、
血小板减少性紫癜、神经症状、肾脏病变等表现及实验室检查的特点,可予以诊断。
鉴别诊断
治疗
治疗:
1.应用激素同时切脾 能使部分病人缓解延长存活时间。
2.激素与
肝素联合应用 也能使部分病例缓解,早期应用
肝素能防止重要器官不可逆性损害。
3.激素与阿司匹林合用 可取得暂时的缓解,阿司匹林能抑制血小板凝聚。
4.镁剂 有报道用镁剂而使本病缓解者,可能镁能竞争抑制钙离子,延缓凝血过程。
5.输血、输血小板及换血 均无明显效果。
预后
预后:本病征预后不良,急性暴发型常在数周内致死,慢性型可持续数月到数年,恶性肿瘤并发者,预后极差。2/3病人可于发病后3个月内死亡,少数病例呈慢性迁延型,转归及病程难以预料。
预防
预防:本病病因未明者占绝大多数,也可继发于其他疾病,同时也发现有家族性发病。常见的继发因素有药物、中毒和免疫疾病等,应积极予以预防,如NO、染料、油漆、蜂毒、蛇毒等,应避免接触造成毒性损害。